
The eccrine sweat glands express muscarinic M3 cholinergic receptors. In an exception to the usual rule that the postganglionic neurotransmitter for the sympathetic nervous system is noradrenaline or adrenaline, the sympathetic nervous system innervates the eccrine sweat glands with cholinergic nerve fibres. Thus, sweating associated with the fight-or-flight response is a sympathetic nervous system response mediated by cholinergic activation of M3 receptors.
But does nicotine not also cause sweating? Nicotine can contribute to sweating in a number of ways. The preganglionic nerve fibres of both the sympathetic and parasympathetic nervous system are cholinergic release acetylcholine to activate nicotinic cholinergic receptors on the ganglionic neurones. Thus nicotine can directly activate the ganglionic neurones triggering activation of the cholinergic postganglionic sympathetic nervous system innervation of the eccrine sweat glands.
The nerve terminals innervating the sweat glands also have presynaptic nicotinic receptors. Application of acetylcholine or nicotine to the skin will activate these nerve terminals triggering action potentials to branches of the nerve innervating adjacent sweat glands to release acetylcholine and activate postsynaptic M3 receptors on these sweat glands. This is referred to as the sudomotor axon reflex.
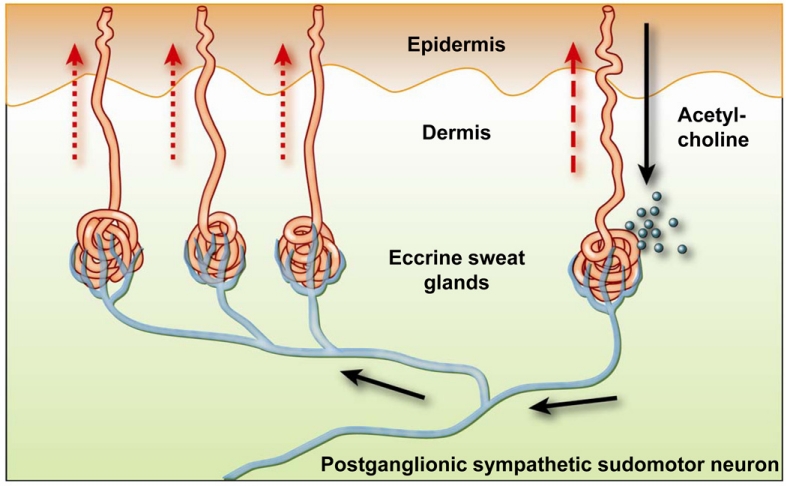 Image credit: http://www.medicavisie.eu/de/technologien/#sudomotor
Image credit: http://www.medicavisie.eu/de/technologien/#sudomotor
Note that to acheive activation of nicotinic receptors exposure to nicotine has to be at a low dose and for a short duration. High doses of nicotine or prolonged exposure to nicotine can lead to depolarising and desensitising block of nicotinic receptor neurotransmission.