
Hot Off the Press: Lysosomal cathepsin D is upregulated in Alzheimer’s disease neocortex and may be a marker for neurofibrillary degeneration
 First authored by postgraduate student Yuek Ling, we report here our latest findings in dementia neurochemistry. Alzheimer’s disease (AD) is characterized by accumulation of β-amyloid plaques (AP) and neurofibrillary tangles (NFT) in the cortex, together with synaptic loss and amyloid angiopathy. Perturbations in the brain lysosomal system, including the cathepsin family of proteases, have been implicated in AD where they may be involved in proteolytic clearance of misfolded and abnormally aggregated peptides. However, the status of cathepsin D (catD) is unclear in Lewy body dementia, the second most common form of neurodegenerative dementia after AD, and characterized by Lewy bodies (LB) containing aggregated α-synuclein. Furthermore, earlier reports of catD changes in AD have not been entirely consistent. We measured CatD immunoreactivities in the temporal (Brodmann area BA21) and parietal (BA40) cortices of well charac
First authored by postgraduate student Yuek Ling, we report here our latest findings in dementia neurochemistry. Alzheimer’s disease (AD) is characterized by accumulation of β-amyloid plaques (AP) and neurofibrillary tangles (NFT) in the cortex, together with synaptic loss and amyloid angiopathy. Perturbations in the brain lysosomal system, including the cathepsin family of proteases, have been implicated in AD where they may be involved in proteolytic clearance of misfolded and abnormally aggregated peptides. However, the status of cathepsin D (catD) is unclear in Lewy body dementia, the second most common form of neurodegenerative dementia after AD, and characterized by Lewy bodies (LB) containing aggregated α-synuclein. Furthermore, earlier reports of catD changes in AD have not been entirely consistent. We measured CatD immunoreactivities in the temporal (Brodmann area BA21) and parietal (BA40) cortices of well charac terized AD brains as well as two clinical subtypes of Lewy body dementia, namely Parkinson disease dementia (PDD) and dementia with Lewy bodies (DLB), known to show varying degrees of concomitant AD pathology. Increased catD immunoreactivities in AD were found for both neocortical regions measured, where they also correlated with neuropathological NFT scores and phosphorylated pSer396 tau burden, and appeared to co-localize at least partly to NFT-containing neurons. In contrast, catD was increased only in BA40 in DLB and not at all in PDD, did not correlate with LB scores, and did not appreciably co-localize with α-synuclein inclusions. Our study suggests that catD upregulation may be an adaptive response to AD-related processes leading to neurofibrillary degeneration, but may not be directly associated with formation of α-synuclein inclusions in Lewy body dementia.
terized AD brains as well as two clinical subtypes of Lewy body dementia, namely Parkinson disease dementia (PDD) and dementia with Lewy bodies (DLB), known to show varying degrees of concomitant AD pathology. Increased catD immunoreactivities in AD were found for both neocortical regions measured, where they also correlated with neuropathological NFT scores and phosphorylated pSer396 tau burden, and appeared to co-localize at least partly to NFT-containing neurons. In contrast, catD was increased only in BA40 in DLB and not at all in PDD, did not correlate with LB scores, and did not appreciably co-localize with α-synuclein inclusions. Our study suggests that catD upregulation may be an adaptive response to AD-related processes leading to neurofibrillary degeneration, but may not be directly associated with formation of α-synuclein inclusions in Lewy body dementia.
Reference
Chai YL, Chong JR, Weng J, et al. (This paper).
 The translocase of the outer membrane (TOM) is a vital mitochondrial transport system facilitating the importation of nuclear encoded proteins into the organelle. While mitochondrial dysfunction, including perturbation of oxidative phosphorylation (OXPHOS) complex, is evident in Alzheimer’s disease (AD), it remains unclear whether the observed OXPHOS deficits may be associated with TOM alterations. In this study, the objective was to correlate TOM subunits with OXPHOS complex proteins in AD. We used postmortem neocortex (BA40) from AD and age-matched controls were processed to obtain mitochondrial enriched homogenates for the measurement of Tom20, Tom22, Tom40, Tom70 as well as components of OXPHOS complex I – V by immunoblotting. We found that Tom20 and Tom70 immunoreactivities were significantly reduced in AD, as were components of OXPHOS complex I and III. Both Tom20 and Tom70 positively correlated with complex III and V, while Tom20 also correlated with complex IV. In conclusion, reductions in certain TOM subunits and their correlations with specific OXPHOS complex proteins suggest that an impaired mitochondrial transportation system may contribute to previously observed oxidative phosphorylation deficits in AD. Follow-up studies are needed to corroborate the present correlative study.
The translocase of the outer membrane (TOM) is a vital mitochondrial transport system facilitating the importation of nuclear encoded proteins into the organelle. While mitochondrial dysfunction, including perturbation of oxidative phosphorylation (OXPHOS) complex, is evident in Alzheimer’s disease (AD), it remains unclear whether the observed OXPHOS deficits may be associated with TOM alterations. In this study, the objective was to correlate TOM subunits with OXPHOS complex proteins in AD. We used postmortem neocortex (BA40) from AD and age-matched controls were processed to obtain mitochondrial enriched homogenates for the measurement of Tom20, Tom22, Tom40, Tom70 as well as components of OXPHOS complex I – V by immunoblotting. We found that Tom20 and Tom70 immunoreactivities were significantly reduced in AD, as were components of OXPHOS complex I and III. Both Tom20 and Tom70 positively correlated with complex III and V, while Tom20 also correlated with complex IV. In conclusion, reductions in certain TOM subunits and their correlations with specific OXPHOS complex proteins suggest that an impaired mitochondrial transportation system may contribute to previously observed oxidative phosphorylation deficits in AD. Follow-up studies are needed to corroborate the present correlative study.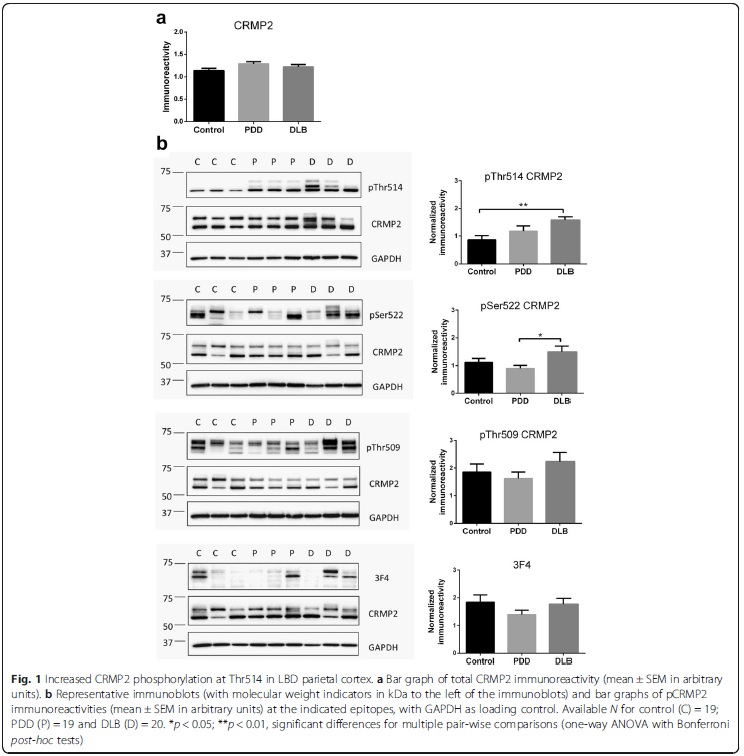 Collapsin response mediator protein-2 (CRMP2) regulates axonal growth cone extension, and increased CRMP2 phosphorylation may lead to axonal degeneration. Axonal and synaptic pathology is an important feature of Lewy body dementias (LBD), but the state of CRMP2 phosphorylation (pCRMP2) as well as its correlations with markers of neurodegeneration have not been studied in these dementias. Hence, we measured CRMP2 phosphorylation at Thr509, Thr514 and Ser522, as well as markers of β-amyloid (Aβ), tau-phosphorylation, α-synuclein and synaptic function in the postmortem neocortex of a longitudinally assessed cohort of LBD patients characterized by low (Parkinson’s disease dementia, PDD) and high (dementia with Lewy bodies, DLB) burden of Alzheimer type pathology. We found specific increases of pCRMP2 at Thr514 in DLB, but not PDD. The increased CRMP2 phosphorylation correlated with fibrillogenic Aβ as well as with losses of markers for axon regeneration (β-III-tubulin) and synaptic integrity (synaptophysin) in LBD. In contrast, pCRMP2 alterations did not correlate with tau-phosphorylation or α-synuclein, and also appear unrelated to immunoreactivities of putative upstream kinases glycogen synthase kinase 3β and cyclin-dependent kinase 5, as well as to protein phosphatase 2A. In conclusion, increased pCRMP2 may underlie the axonal pathology of DLB, and may be a novel therapeutic target. However, antecedent signaling events as well as the nature of pCRMP2 association with Aβ and other neuropathologic markers require further study.
Collapsin response mediator protein-2 (CRMP2) regulates axonal growth cone extension, and increased CRMP2 phosphorylation may lead to axonal degeneration. Axonal and synaptic pathology is an important feature of Lewy body dementias (LBD), but the state of CRMP2 phosphorylation (pCRMP2) as well as its correlations with markers of neurodegeneration have not been studied in these dementias. Hence, we measured CRMP2 phosphorylation at Thr509, Thr514 and Ser522, as well as markers of β-amyloid (Aβ), tau-phosphorylation, α-synuclein and synaptic function in the postmortem neocortex of a longitudinally assessed cohort of LBD patients characterized by low (Parkinson’s disease dementia, PDD) and high (dementia with Lewy bodies, DLB) burden of Alzheimer type pathology. We found specific increases of pCRMP2 at Thr514 in DLB, but not PDD. The increased CRMP2 phosphorylation correlated with fibrillogenic Aβ as well as with losses of markers for axon regeneration (β-III-tubulin) and synaptic integrity (synaptophysin) in LBD. In contrast, pCRMP2 alterations did not correlate with tau-phosphorylation or α-synuclein, and also appear unrelated to immunoreactivities of putative upstream kinases glycogen synthase kinase 3β and cyclin-dependent kinase 5, as well as to protein phosphatase 2A. In conclusion, increased pCRMP2 may underlie the axonal pathology of DLB, and may be a novel therapeutic target. However, antecedent signaling events as well as the nature of pCRMP2 association with Aβ and other neuropathologic markers require further study.

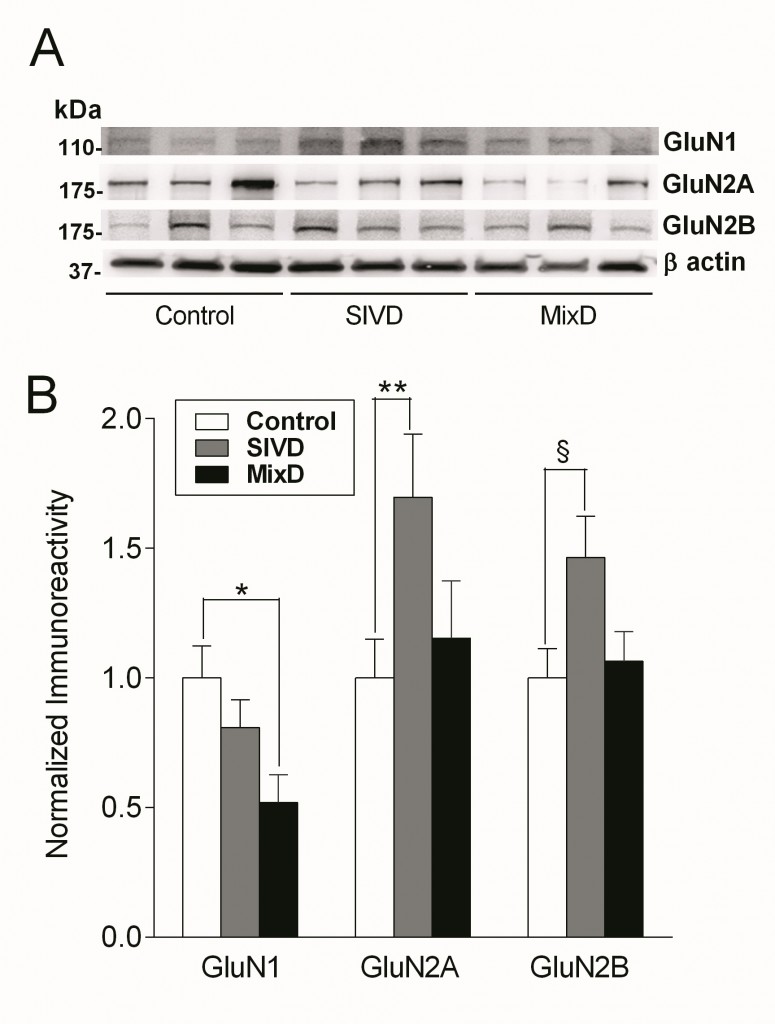 Glutamatergic deficits are well-established neurochemical findings in Alzheimer’s disease (AD) and are thought to underlie both cognitive and behavioral symptoms of the disease. However, it is unclear whether subcortical ischemic vascular dementia (SIVD) and mixed SIVD/AD (MixD) manifest similar changes in the glutamatergic system. In this study, we measured the immunoreactivities of NMDA receptor GluN1, GluN2A, and GluN2B subunits in SIVD and MixD in postmortem neocortical tissues from a cohort of well-characterized, longitudinally followed-up patients with SIVD and MixD, together with age-matched controls. We found a significant reduction of GluN1 only in MixD, while significant increases of GluN2A and GluN2B were found only in SIVD. Furthermore, GluN1 loss and GluN2A/2B upregulation was associated respectively with higher Braak stages and lacunar infarct scores. Our data therefore suggest that the differential alterations of GluN subunits in SIVD and MixD may result from separate, interacting disease processes, and point to the potential utility of glutamatergic approaches for pharmacotherapy.
Glutamatergic deficits are well-established neurochemical findings in Alzheimer’s disease (AD) and are thought to underlie both cognitive and behavioral symptoms of the disease. However, it is unclear whether subcortical ischemic vascular dementia (SIVD) and mixed SIVD/AD (MixD) manifest similar changes in the glutamatergic system. In this study, we measured the immunoreactivities of NMDA receptor GluN1, GluN2A, and GluN2B subunits in SIVD and MixD in postmortem neocortical tissues from a cohort of well-characterized, longitudinally followed-up patients with SIVD and MixD, together with age-matched controls. We found a significant reduction of GluN1 only in MixD, while significant increases of GluN2A and GluN2B were found only in SIVD. Furthermore, GluN1 loss and GluN2A/2B upregulation was associated respectively with higher Braak stages and lacunar infarct scores. Our data therefore suggest that the differential alterations of GluN subunits in SIVD and MixD may result from separate, interacting disease processes, and point to the potential utility of glutamatergic approaches for pharmacotherapy.

 Dementia with Lewy bodies and Parkinson’s disease dementia are different clinical phenotypes of Lewy body dementias (LBD) differentiated by the temporal relationship between parkinsonism and dementia onset. At present, it is unclear whether the glutamatergic system is affected in these disorders. In this study, we measured α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor GluA subunits in the postmortem neocortex of a cohort of prospectively studied Lewy body dementia cases, as well as age-matched controls by immunoblotting. We found moderate losses of GluA2/3/4 immunoreactivities in LBD which correlated with higher predeath Hoehn & Yahr scores, a measure of motor disabilities; but not with dementia severity, cortical Lewy body burden, or amyloid plaque and neurofibrillary tangle burden. Our study suggests that GluA2/3/4 losses may be a neurochemical marker of motor disability in Lewy body dementias. Given recent proposals to use AMPA receptor antagonists in Parkinson’s disease, especially for levodopa-induced dyskinesia (Johnson et al. 2009), our data suggest both potential and caution in extending such therapeutic rationales to LBD in view of altered levels of receptor drug targets.
Dementia with Lewy bodies and Parkinson’s disease dementia are different clinical phenotypes of Lewy body dementias (LBD) differentiated by the temporal relationship between parkinsonism and dementia onset. At present, it is unclear whether the glutamatergic system is affected in these disorders. In this study, we measured α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor GluA subunits in the postmortem neocortex of a cohort of prospectively studied Lewy body dementia cases, as well as age-matched controls by immunoblotting. We found moderate losses of GluA2/3/4 immunoreactivities in LBD which correlated with higher predeath Hoehn & Yahr scores, a measure of motor disabilities; but not with dementia severity, cortical Lewy body burden, or amyloid plaque and neurofibrillary tangle burden. Our study suggests that GluA2/3/4 losses may be a neurochemical marker of motor disability in Lewy body dementias. Given recent proposals to use AMPA receptor antagonists in Parkinson’s disease, especially for levodopa-induced dyskinesia (Johnson et al. 2009), our data suggest both potential and caution in extending such therapeutic rationales to LBD in view of altered levels of receptor drug targets.